miRNAs - Work in Progress
Introduction
MicroRNAs (miRNAs) represent one of the most extensively studied classes of post-transcriptional gene regulators, yet the mechanistic details of their biogenesis have only recently been fully elucidated. A critical distinction in the field concerns the transcription machinery responsible for generating primary miRNA transcripts (pri-miRNAs). While many organisms produce miRNAs via RNA polymerase III (Pol III) transcription, the canonical pathway in mammalian cells proceeds through RNA polymerase II (Pol II), the same enzyme responsible for mRNA transcription. This distinction has profound implications for how synthetic miRNA pathways can be engineered.
When transcribed by Pol II, pri-miRNAs acquire the hallmarks of standard mRNAs: a 5' 7-methylguanosine cap and a 3' polyadenylate tail. The genomic context of mammalian miRNAs is remarkably diverse. Approximately 50–70% of miRNAs reside within introns of protein-coding or non-coding genes, allowing their processing and expression to be coupled with host gene transcription. The remainder are distributed across intergenic regions or embedded within exons and untranslated regions (UTRs). This architectural flexibility has enabled the development of sophisticated synthetic approaches to miRNA expression and function.
The miR-E system, developed by Fellmann and colleagues in 2013, represents a milestone in rational engineering of synthetic miRNA scaffolds. By anchoring a synthetic miRNA sequence within a standardized Pol II expression cassette, the miR-E approach provides a generalizable framework for robust and reliable miRNA production. This review examines the design principles, processing steps, and functional advantages of the miR-E scaffold, with particular emphasis on how sequence determinants at multiple processing checkpoints coordinate to maximize mature miRNA yield.
The miR-E 97-mer Scaffold
The core of the miR-E system is a 97-nucleotide DNA oligonucleotide, typically synthesized using IDT Ultramers or similar high-fidelity synthesis platforms. This 97-mer represents the minimal sequence required to encode a functional miRNA within the context of Pol II transcription. The oligo is initially amplified by PCR to generate sufficient material for cloning, then inserted into a recipient expression vector at defined restriction sites, typically XhoI and EcoRI, flanking the cloning site.
The critical insight underlying the miR-E design is that the 97-mer alone is insufficient for efficient Drosha processing. The Drosha/DGCR8 Microprocessor complex requires ~125 nucleotides of flanking sequence on each side of the ~70 nucleotide pre-miRNA substrate for accurate recognition and cleavage. These flanking regions provide structural context that the enzyme uses to identify the substrate-enzyme interface. The recipient vector—typically containing complementary DNA (cDNA) encoding a marker protein such as GFP upstream of the miR-30 insertion site—provides precisely this required context.
Within the 97-mer itself, several invariant sequences have been carefully preserved to ensure faithful processing. The 5' flanking region comprises a 19-nucleotide sequence that forms the basal stem of the pri-miRNA structure, helping to nucleate the secondary structure recognized by Drosha. The central 97-mer contains the native mir-30a terminal loop sequence, which serves as a structural anchor and is ultimately removed during Dicer processing. The 3' flanking region consists of a 17-nucleotide sequence that concludes with a critically restored CNNC trinucleotide-plus-C motif. This tetranucleoside motif, as discussed below, is recognized by the SR protein SRp20 and directly influences Microprocessor recruitment efficiency.

Step-by-Step Processing
The journey from transcribed pri-miRNA to functional mature miRNA involves a precisely orchestrated series of enzymatic steps, each with distinct sequence recognition requirements and structural constraints.
Step 1: Pol II Transcription and pri-miRNA Formation. The process begins when Pol II encounters the integrated expression cassette and initiates transcription upstream of the marker gene. Transcription proceeds through the GFP coding sequence, then through the miR-30 flanking context and the 97-mer core, continuing until the natural or engineered termination signal is reached. The complete transcript—typically 2–4 kilobases in length—constitutes the pri-miRNA. This molecule bears all the hallmarks of an mRNA: a 5' cap added co-transcriptionally, and a 3' polyadenylate tail added post-transcriptionally by cleavage and polyadenylation machinery. The pri-miRNA is exported from the nucleus (via processes described below) before Drosha processing, unlike the case with some intronic miRNAs, which can undergo Drosha cleavage in the nucleus while still linked to unspliced introns.
Step 2: Drosha/DGCR8 Cleavage to pre-miRNA. The Microprocessor complex, a ribonucleoprotein assembly centered on the RNase III enzyme Drosha and its partner DGCR8, cleaves the pri-miRNA ~11 base pairs away from the junction between the basal stem and the stem proper. This cleavage produces a ~65 nucleotide hairpin termed the precursor miRNA (pre-miRNA), which retains a characteristic 2-nucleotide 3' overhang—a signature feature of RNase III enzymes. The DGCR8 partner protein functions as a molecular ruler, using its architecture to position Drosha at the correct distance from the substrate terminus. Notably, when the miRNA is embedded within an mRNA (as in the miR-E system), the 5' and 3' products flanking the miRNA can have different fates. The GFP mRNA sequence upstream of the miR-30 flanking region often remains partially functional, particularly if the Drosha cleavage point occurs downstream of the GFP stop codon—although excess unstructured RNA will be targeted for degradation by nuclear exonucleases. The 3' flanking RNA product is rapidly degraded by nuclear exonucleases, a process that is mechanistically distinct from and more efficient than the decay of the 5' product.
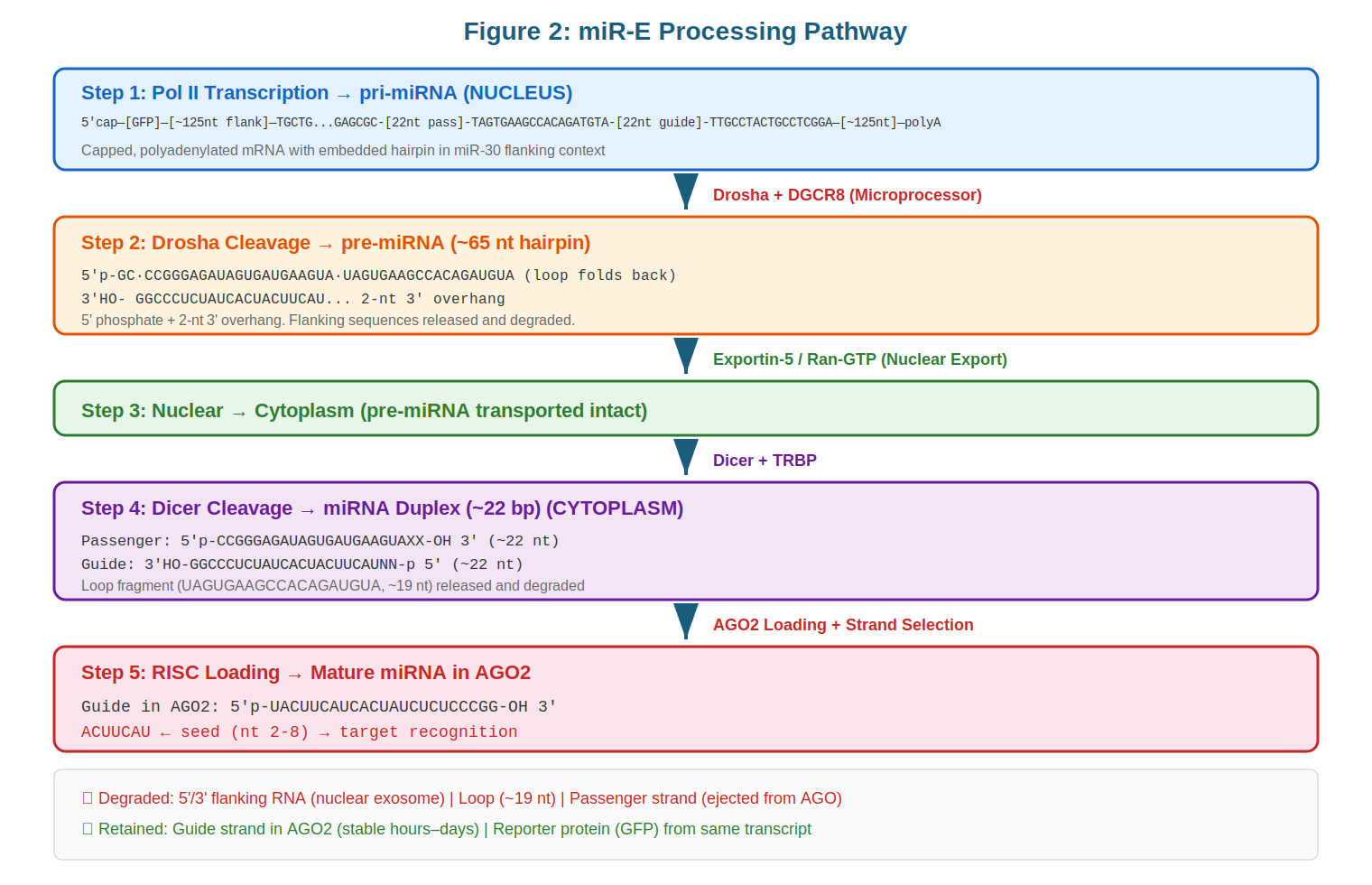
Step 3: Exportin-5 Nuclear Export. The ~65 nt pre-miRNA bearing a 2-nucleotide 3' overhang is recognized by the nuclear export receptor Exportin-5 in a complex with the Ran GTPase bound to GTP. Exportin-5 has two key sequence recognition requirements: the 2-nucleotide 3' overhang and a stem of at least 16 base pairs. These features, present in all natural pre-miRNAs and maintained in the miR-E design, ensure that only properly processed pre-miRNAs—not pri-miRNA or other small RNA species—are transported to the cytoplasm. This export step is rate-limiting in Pol III-based systems (such as shRNAs), where high transcription rates can saturate Exportin-5 capacity and trigger cellular stress responses.
Step 4: Dicer/TRBP Cleavage to miRNA Duplex. Once in the cytoplasm, Dicer, another RNase III enzyme, cleaves the pre-miRNA near its terminal loop. Dicer measures approximately 22 nucleotides from the 5' phosphate (corresponding to the 5' end of the pre-miRNA hairpin) and performs a cut that removes the terminal loop while simultaneously releasing the opposite end of the duplex. This produces a ~22 nucleotide miRNA duplex with 2-nucleotide 3' overhangs on both the 5' and 3' strands. The terminal loop, which contains conserved sequences but is no longer needed for gene silencing, is rapidly degraded by exonucleases.
Step 5: RISC Assembly and Strand Selection. The duplex miRNA is loaded onto Argonaute (AGO) proteins, the catalytic core of the RNA-induced silencing complex (RISC). In this context, strand selection—the process by which one strand is retained as the guide strand and the other is degraded—becomes critical. Natural miRNAs often have asymmetric base pairing near their 5' ends, with the guide strand (the one that will be retained) being thermodynamically less stable at this position. This asymmetry enables the RISC machinery to preferentially unwind and load the correct strand. In the miR-E design, the synthetic miRNA sequence is engineered to contain deliberate mismatches that enforce this thermodynamic asymmetry, ensuring that the antisense strand—designed to target mRNAs of interest—is preferentially loaded into RISC. Once loaded, the guide strand is extraordinarily stable, with residence times measured in hours to days. In contrast, the passenger strand (the sense strand) is typically ejected from RISC within seconds and rapidly degraded.
The 2-nucleotide 3' overhang produced by RNase III enzymes is not merely a byproduct—it serves as a molecular signature that gates progress through the miRNA biogenesis pathway, allowing the cell to distinguish properly processed intermediates from spurious RNAs.
Fate of Processing Intermediates
A complete understanding of miRNA biogenesis requires accounting for the fates of all molecular species generated during processing. The 5' flanking RNA, which in the miR-E system consists of the GFP mRNA plus the 5' miR-30 context, is partially retained as functional GFP mRNA—provided the Drosha cleavage point is downstream of the GFP coding sequence and stop codon. Any excess GFP mRNA that is not translated is subject to degradation by the nuclear exosome and cytoplasmic decay pathways. The 3' flanking RNA generated by Drosha cleavage is composed of the 3' miR-30 context plus vector sequences and is invariably degraded by 5'→3' exonucleases (particularly the exosome) within the nucleus. The terminal loop removed by Dicer is a short oligonucleotide (typically 4–6 nucleotides) and is degraded rapidly and non-specifically by cellular nucleases. The passenger strand ejected during RISC loading is targeted for rapid degradation, with half-lives measured in seconds, through mechanisms that may involve both exonuclease attack and endonucleolytic cleavage by residual RISC complexes. The mature guide strand, by contrast, is stabilized by its incorporation into AGO-RISC and can persist for hours to days, enabling sustained and efficient gene silencing.
miR-E Versus Classic shRNA Systems
To appreciate the innovation of the miR-E approach, it is instructive to compare it with earlier synthetic small RNA strategies, particularly those based on short hairpin RNAs (shRNAs). Classic shRNA systems rely on Pol III transcription, typically under the control of the U6 or H1 promoter. These promoters generate short (~100–300 nucleotide), un-capped, un-polyadenylated transcripts that lack the structural context recognized by Drosha. Consequently, classic shRNAs bypass Drosha entirely and enter the biogenesis pathway at the level of Exportin-5 nuclear export, being recognized by that export factor based solely on their 2-nucleotide overhang and stem length.

This Drosha-bypass strategy has several consequences. First, because shRNAs lack upstream flanking sequence, they are recognized by Dicer without the benefit of the architectural constraints that normally guide RISC loading decisions. Many shRNAs are designed to be perfectly complementary across their full length, which paradoxically leads to inefficient strand selection: both the sense and antisense strands have equal thermodynamic stability and are loaded into RISC at comparable rates. This contrasts with natural miRNAs, where the guide strand is strongly preferred. Second, Pol III transcription is inherently very efficient, often proceeding at 10–100 fold higher rates than Pol II transcription of comparable sequences. When shRNA transgenes are integrated into the genome, this high transcription rate can saturate the Exportin-5/Ran-GTP export machinery. The resulting accumulation of pre-shRNA in the nucleus triggers stress responses and can lead to cellular toxicity, as documented in multiple studies. Third, the combination of high expression and dual-strand loading can overwhelm RISC capacity, further exacerbating toxicity.
The miR-E system addresses each of these limitations. By anchoring the miRNA within a Pol II expression cassette, miR-E achieves modestly higher transcript levels than typical Pol II-driven mRNAs but substantially lower levels than Pol III shRNA systems, avoiding saturation of export machinery. By maintaining the full canonical processing pathway—including Drosha cleavage—miR-E benefits from the structural constraints that ensure single-strand loading of the guide strand. Finally, by incorporating engineered mismatches to enforce strand selection, miR-E mimics the thermodynamic asymmetry of natural miRNAs, further ensuring that only the desired strand is loaded. The result is a system that combines the reliability of synthetic RNA scaffolds with the efficiency and specificity of natural miRNA biogenesis.
The Key Innovation: The CNNC Motif
A particularly instructive example of sequence determinism in miRNA processing emerged from detailed mechanistic studies of synthetic Drosha substrates. In early work designing synthetic miRNA scaffolds, researchers built expression constructs based on the mir-30 backbone but found that the efficiency of mature miRNA production was far below theoretical expectations—typically yielding only 10–20% of the small RNA levels achieved with optimized natural miRNAs.
The source of this inefficiency was traced to an inadvertent disruption of a conserved sequence motif. In the original synthetic scaffold, an EcoRI restriction site (sequence GAATTC) was incorporated into the 3' flanking region for cloning purposes. This site, while useful for molecular cloning, had the unintended consequence of destroying a highly conserved CNNC trinucleotide (where N represents any nucleotide) followed by a cytidine at positions +17 through +20 relative to the Drosha cleavage site on the 3' side of the pre-miRNA.
The CNNC motif is recognized by SRp20 (also known as SRSF3), a member of the serine/arginine-rich (SR) family of RNA-binding proteins. SRp20 binding to this motif is essential for efficient recruitment of the Microprocessor complex to pri-miRNA substrates. By disrupting the CNNC motif, the EcoRI site indirectly weakened Microprocessor binding, dramatically reducing Drosha cleavage efficiency. When Fellmann and colleagues relocated the EcoRI site to a position that preserved the wild-type CNNC motif, mature miRNA production increased ~10-fold, bringing synthetic constructs into line with natural miRNA biogenesis rates.
This finding has broader implications. It demonstrates that synthetic RNA engineering cannot proceed solely from considerations of minimum structural requirements; rather, biological sequences contain multiple overlapping functional elements whose disruption can have pleiotropic effects. The CNNC motif example also highlights the importance of understanding the full cast of factors required for efficient processing—in this case, revealing an unexpected role for the SR protein SRp20 in Microprocessor recruitment. Today, the CNNC motif is carefully preserved in all optimized synthetic miRNA scaffolds, and synthetic designs increasingly incorporate other known sequence determinants (such as the basal stem length and loop stability) to maximize processing efficiency.
Conclusion
The miR-E scaffold represents a synthesis of decades of miRNA research into a practical tool for synthetic biology and functional genomics. By marrying the advantages of Pol II-driven expression (which provides structural context and moderate expression levels) with the rational engineering of sequence determinants (which optimize processing efficiency and strand selection), the miR-E approach achieves high yields of functional, precisely designed miRNAs. Understanding the detailed mechanisms of miRNA biogenesis—from pri-miRNA transcription through Drosha cleavage, nuclear export, Dicer processing, and RISC loading—provides a foundation for further improvements in synthetic small RNA design and for a deeper mechanistic understanding of post-transcriptional gene regulation in mammalian cells.
Sources
- Fellmann, C., Hoffmann, T., Sridhar, V., et al. (2013). An optimized microRNA backbone for effective single-copy RNAi. Cell Reports. https://doi.org/10.1016/j.celrep.2013.11.020
- Lee, Y., Kim, M., Han, J., et al. (2004). MicroRNA genes are transcribed by RNA polymerase II. EMBO Journal, 23(20), 4051–4060.
- Auyeung, V. C., Ulitsky, I., McGeary, S. E., & Bartel, D. P. (2013). Beyond secondary structure: primary-sequence determinants license 3′ UTRs for cytoplasmic localization and regulatory cross-talk. Cell, 154(2), 385–398. https://doi.org/10.1016/j.cell.2013.01.031
- Han, J., Lee, Y., Yeom, K.-H., et al. (2006). Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell, 125(5), 887–901.
- McBride, J. L., Boudreau, R. L., Harper, S. Q., et al. (2008). Artificial miRNAs mitigate shRNA toxicity and improve therapeutic efficacy. Proceedings of the National Academy of Sciences USA, 105(15), 5868–5873.