Lentivirus - Work in Progress
Domesticating HIV
Frequently employed for gene delivery to cells, lentivirus is a derivative of human immunodeficiency virus type 1 (HIV-1). Using a deadly pathogen for everyday research may appear counterintuitive, but HIV-1’s daunting characteristics (stable genomic integration and efficacious transduction of non-dividing cells) are precisely why it’s useful. Retaining these delivery capabilities while eliminating replication competency was progressively achieved over four generations of packaging systems (Fig. 1).
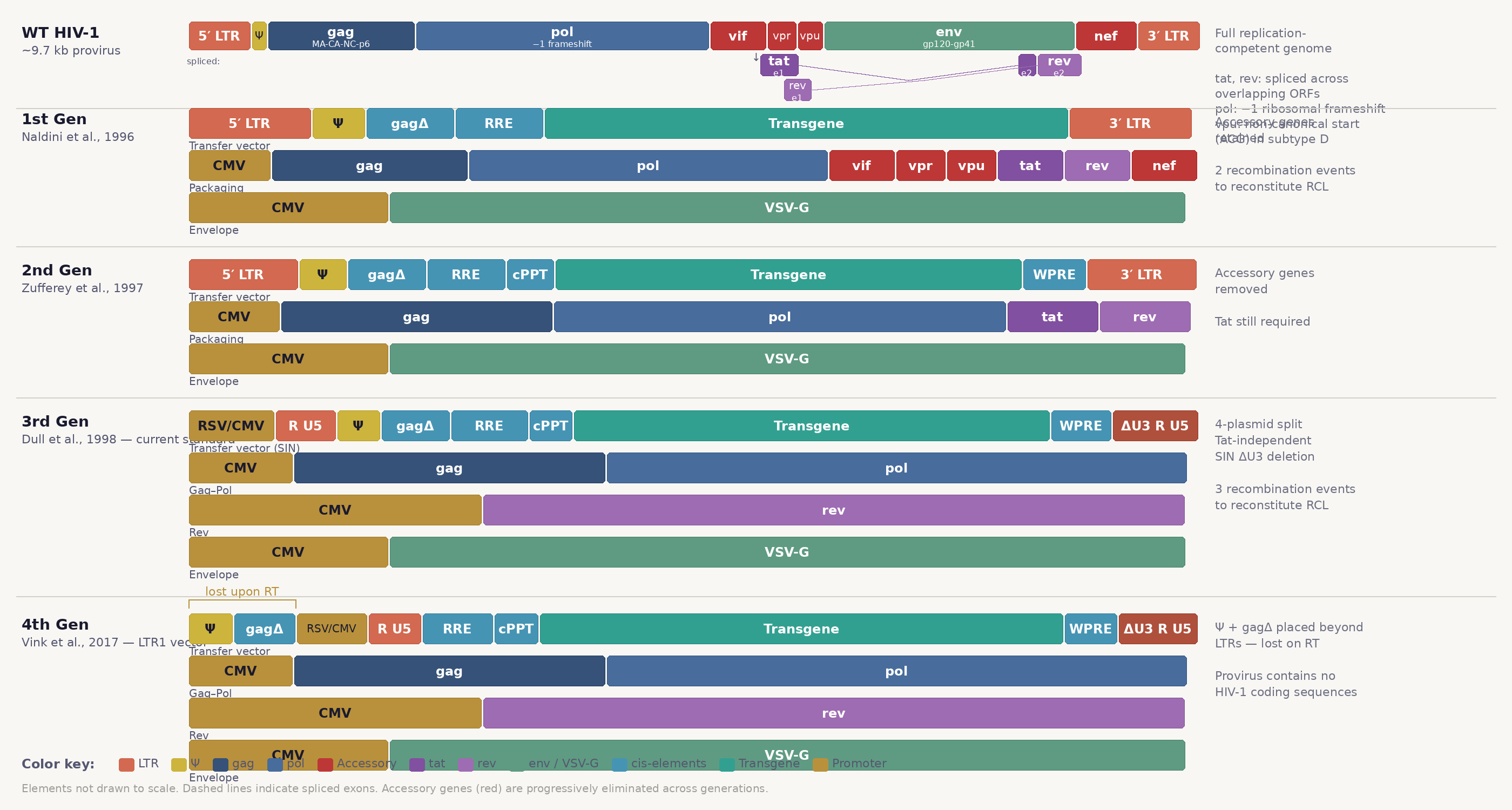
Figure 1. Progressive domestication of the HIV-1 genome across lentiviral vector generations.
Like many viruses, wild-type (wt) HIV-1 is remarkably efficient: its ~9.7 kb (kilobase) genome encodes nine open reading frames (ORFs). HIV-1’s ORFs fall into three categories. Structural and enzymatic genes are essential for particle assembly and replication: gag encodes the polyprotein precursor (MA–CA–NC–p6) that forms the MAtrix, CApsid, and NucleoCapsid; pol encodes a protease, reverse transcriptase, and integrase (expressed as a Gag-Pol fusion with pol translated via a −1 ribosomal frameshift); and env encodes surface (gp120) and transmembrane (gp41) glycoproteins responsible for receptor binding and membrane fusion. The two regulatory genes, tat and rev, are imperative for viral gene expression: Tat binds the trans-activation response element (TAR) stem-loop to enhance transcription driven by the 5′ LTR promoter while Rev binds the Rev response element (RRE) to export viral mRNA from the nucleus. Tat and Rev are essential for productive infection but can be supplied in trans or (in the case of Tat) functionally replaced entirely by an exogenous promoter (commonly CMV or RSV).
The accessory genes (vif, vpr, vpu, nef) are dispensable for replication in permissive cell lines but crucial for in vivo wt HIV-1 pathogenesis36. Vif (Virion Infectivity Factor) targets APOBEC3G, a cytidine deaminase which lethally mutates G→A in viral cDNA, for proteasomal degradation via a CUL5–ElonginB/C E3 ubiquitin ligase complex37. Vpr (Viral Protein R) arrests cell cycle via the SLX4 endonuclease complex and facilitates nuclear import of the pre-integration complex in macrophages38. Vpu (Viral Protein UVpuvpu also shares a bicistronic mRNA with env and initiates from a non-canonical ACG start codon in subtype D isolates. There are nine common HIV-1 subtypes, with subtype C comprising approximately 50% of global cases.) degrades CD4CD4CD4 is the primary receptor for wt HIV-1 entry; gp120 binding exposes a co-receptor site, and engagement of CCR5 or CXCR4 triggers gp41-mediated membrane fusion. CCR5 is the same gene disrupted by He Jiankui in the 2018 germline-editing experiment on twin embryos, an attempt to confer HIV resistance based on the naturally occurring CCR5-Δ32 deletion found in ~1% of Europeans41. (the HIV-1 primary receptor) and antagonizes BST-2/tetherin, which traps budding virions on the cell surface39. Nef (Negative Factor, a misnomer :) helps downregulate CD4, removes MHC-I from the cell surface to evade T-cell recognition, and enhances virion infectivity by debated mechanisms40. Vpu- and Nef-mediated CD4 downregulation prevent re-entry of progeny virions (superinfection) which is cytotoxic and wasteful. As these four proteins aren’t requisite for vector production, their removal in second-generation systems retains titer while improving biosafety.
First-generation systems1 partitioned the viral genome into two plasmids: a transfer vector retaining cis-acting elements for RNA processing, packaging, reverse transcription, and integration; and a packaging construct supplying trans-acting proteins (Gag, Pol, Tat, Rev) except Env, which was replaced by VSV-G on a separate plasmid. This eliminated replication-competent lentivirus from a single recombination event, but retained perilous accessory genes (vif, vpr, vpu, nef). Second-generation systems2 stripped accessory genes away but kept Tat and the wt 5′ LTR: a three-plasmid system (transfer, packaging, envelope). Many labs still use second-generation systems.
Third-generation systems3 eliminated Tat dependence by replacing the U3 region of the 5′ LTR with a constitutive promoter (typically CMV or RSV) and separated Rev onto its own plasmid, producing the currently standardized four-plasmid architecture (transfer vector, Gag-Pol, Rev, and envelope; Fig. 1 & 2). This split requires three independent recombination events to reconstitute anything resembling replication-competent HIV-1. The self-inactivating (SIN) deletion in the 3′ LTR U3 region, copied to both LTRs upon reverse transcription, provides a final safety layer by silencing the proviral promoter in transduced cells. The plasmid split can reduce titers, but the deficit is protocol-dependent and can be recovered by swapping the chimeric 5′ LTR promoter (e.g. from RSV to CMV) and optimizing plasmid ratios35. Each of the system’s components (the cis-acting elements of the transfer vector, the Gag-Pol packaging machinery, Rev, and the VSV-G envelope) are described below.
Worth mention, Vink et al. (2017)31 developed the fourth-generation LTR1 vector, which eliminates HIV-1 packaging sequences from the integrated provirus entirely by preventing reverse transcription of Ψ and gag due to their placement outside the LTRs (which skips the first strand transfer required for reverse-transcription). The resultant provirus contains minimal HIV-1 sequences (e.g. minimal LTR att sites), reducing theoretical mobilization risk to near zero and accelerating transgene expression by removing inhibitory RNA structures from the 5′ UTR. These plasmids haven’t seen adoption (second- and third-generation are standard), but represent a continued trajectory of domestication.
After three decades, a modern third-generation transfer vector retains ~8% of the original HIV-1 genome, exclusively cis-acting regulatory sequences, while all protein-coding functions are supplied from separate, unpackageable helper plasmids. Understanding these elements in detail is essential for custom lentiviral packaging systems.
The Transfer Plasmid

Figure 2. RNA secondary structure of a third-generation lentiviral transfer vector (ViennaRNA MFE fold, naview layout). Nucleotide circles are colored by cis-acting region: TAR (nt 1–57), poly(A) hairpin (nt 58–104), U5 (nt 105–181), PBS (nt 182–242), SL1/DIS (nt 243–277), SL2/SD (nt 278–296), SL3/Ψ (nt 297–310), SL4/AUG (nt 311–341), partial gag (nt 342–631), RRE (nt 632–862), cPPT/CTS (nt 863–980), and 3′ ΔU3-R-U5 LTR (nt 981–1160). Brackets indicate the core packaging signal (Ψ, SL1–SL3), the extended packaging signal (broad Ψ, SL1–SL4 + gag), and the NC zinc-knuckle binding region (SL2–SL3). Transgene and WPRE sequences are omitted (dashed).
The 5′ UTR and packaging signal (Ψ). The 5′ UTR is functionally dense, spanning from the transcription start site (R in the 5′ LTR) through the first ~350 nucleotides. It contains structured RNA elements: TAR, a polyadenylation signal (which is actively suppressed to prevent premature termination), PBS, and stem-loops 1–4. These elements govern RNA processing, nuclear export, dimerization, and selective packaging into virions. Third-generation systems replaced U3 in the 5′ LTR with an exogenous Pol II promoter, driving expression starting from HIV-1 R (~100 nt) and U5 (~80 nt). R contains TAR and the polyadenylation signal, providing sequence complementarity for first strand transfer. U5 contains the 5′ att site recognized by integrase. Both are essential for reverse transcription and integration.
TAR (Trans-Activation Response element). The first ~60 nucleotides of the transcript form a stable stem-loop within R. In wt HIV-1, Tat binds the bulge of TAR to recruit P-TEFb (CDK9/Cyclin T1), which phosphorylates the Pol II CTD to enhance transcriptional elongation. In third-generation vectors, an exogenous Pol II promoter renders TAR unneeded for transcription. Still, TAR is retained as it lies within R, and R–R sequence complementarity is required for the first strand transfer.
The polyadenylation signal (PAS). The AAUAAA hexamer and its downstream GU-rich element reside within R and are present in both LTRs. At the 5′ end, premature cleavage and polyadenylation are suppressed, or the transcript would be truncated to ~600 nt. This suppression is achieved in three ways: the poly(A) signal hairpin’s secondary structure sequesters the motif from CPSF recognition; the proximity of the 5′ cap sterically inhibits 3′ processing machinery; and U1 snRNP binding the downstream splice donor in SL2 actively represses the polyadenylation complex. The 3′ LTR lacks these elements: its PAS is fully accessible, and CPSF/CstF recognize and cleave the transcript ~20 nt downstream.
tRNALys3 anneals to the PBS, providing the 3′-OH primer for RT
The primer binding site (PBS). An 18-nucleotide sequence immediately downstream of U5, complementary to the 3′ end of human tRNALys3. The host cell’s tRNA binds the PBS and acts as the primer for minus-strand DNA synthesis. Its 5′ placement is why the first strand transfer is obligatory: RT synthesizes DNA 5′→3′, reading through U5 and R until nearly the 5′ end of the genomic RNA. Lacking a primer site near the 3′ LTR, the only way to copy the remaining genome is for the nascent cDNA to jump to the 3′ R sequence and continue synthesis from there. Notably, fourth-generation systems that relocate the PBS downstream of the 3′ LTR require just one strand transfer, whereas third-generation systems and wt HIV-1 require two.
DIS kissing-loop: two SL1 palindromes initiate RNA dimerization
Kissing loops
HIV-1 DIS kissing-loops have been programmably co-opted to form condensates of RNA nanostars annealed to one another. Fabrini et al. (2024, Nature Nanotechnology 19, 1665–1673) designed four-armed RNA nanostars whose arms terminated in kissing-loop sequences derived from the HIV-1 DIS, and visualized micron-scale RNA condensate formation via fluorescent light-up aptamers (like Broccoli) that fluoresce upon interaction with small-molecule fluorophore mimics.
SL1 / Dimerization Initiation Site (DIS). SL1 contains a 6-nucleotide palindromic loop (e.g. GCGCGC in HIV-1 subtype B) that mediates the kissing-loop interaction initiating RNA dimerization. Two genomic RNAs with complementary DIS sequences form a kissing complex that matures into an extended duplex via Gag’s NC domain.
SL2 (major splice donor). SL2 contains the major splice donor site (SD1). In wild-type HIV-1, this 5′ splice site generates over 40 alternatively spliced isoforms encoding Tat, Rev, Nef, Vif, Vpr, Vpu, and Env. In transfer vectors, all downstream splice acceptors are deleted along with the coding sequences they access, so productive splicing is largely non-functional. SL2 is retained because it contributes to packaging efficiency as part of the extended Ψ signal, and because U1 snRNP binding at the SD helps suppress premature polyadenylation. Generally, cis-acting introns within transfer vectors are spliced out and don’t reduce packaging.
SL3 (core Ψ). SL3 presents a GGAG tetraloop and flanking stem bound by the zinc knuckle domains of the NC region of Gag. It is one of the main determinants (alongside SL1 and the 5′ gag region) that bias Gag toward vector RNA over the rest of the producer-cell RNA pool. Mutations in the GGAG tetraloop reduce packaging efficiency 10–100-fold in reporter assays, though the exact selectivity in cellulo is harder to pin down because Gag also contacts RNA non-specifically via NC and MA.
SL4 and the gag fragment (gag Δ). SL4 encompasses the gag AUG start codon, which is mutated (typically ATG→CTG) to prevent Gag translation from vector RNA. Downstream of SL4, ~360 bp of the 5′ gag coding sequence is included as part of the extended packaging signal, and packaging efficiency drops substantially without it as the NC zinc knuckles interact with sequences well into gag. Mutating the start codon prevents ribosomes from translating non-functional and potentially harmful truncated Gag peptides from vector RNA, while leaving the surrounding gag sequence available for its packaging role. Structural work by Vamva et al. (2022, Nucleic Acids Research) revealed a U5–gag interaction that contributes to the monomer–dimer RNA equilibrium involved in packaging.
Central polypurine tract and central termination sequence (cPPT/CTS). Derived from HIV-1 pol, the cPPT is a second purine-rich, RNase H-resistant sequence that primes an internal plus-strand DNA synthesis during reverse transcription. When this internal plus strand reaches the plus strand initiated from the upstream 3′ PPT after the second strand transfer, RT displaces a short stretch of the downstream plus strand before terminating at the CTS (a sequence that makes RT pause and dissociate), creating a ~100 nt triple-stranded “central DNA flap.” This flap is not fully resolved until after nuclear import, and including the cPPT/CTS in vector designs typically boosts transduction ~5–10-fold11. The underlying mechanism is still debated: proposals include facilitating nuclear import of the pre-integration complex, promoting uncoating, or improving the completion of plus-strand synthesis itself, and the relative contributions of these steps have not been fully resolved.
Internal promoter and transgene cassette. This region carries the desired cargo (e.g. perturbations, barcodes, selection markers). Homology within these elements can drive recombination within a vector and across vectors in pooled libraries.
The Rev Response Element (RRE). The RRE is an ~350 nt highly structured RNA element derived from HIV-1 env. Rev binds as a monomer to the high-affinity site on stem-loop IIB of the RRE, then oligomerizes cooperatively along the RNA to form a multimeric Rev–RRE ribonucleoprotein complex. This complex engages the CRM1/Exportin-1 nuclear export receptor via Rev’s leucine-rich nuclear export signal (NES), mediating transport of the unspliced vector RNA through the nuclear pore. Without Rev–RRE, unspliced HIV-1 transcripts are retained in the nucleus and degraded, a pathway enforced by host RNA surveillance mechanisms to prevent export of incompletely spliced pre-mRNAs.
WPRE (Woodchuck Hepatitis Virus Posttranscriptional Regulatory Element). WPRE is a ~600 nt element from woodchuck hepatitis virus that can post-transcriptionally stabilize RNAs. It is almost always included, albeit not required, upstream of the 3′ LTR.
The 3′ polypurine tract (3′ PPT). The 3′ PPT is an RNase H-resistant purine-rich sequence (~15 nt, nearly all A/G) located immediately upstream of U3 in the 3′ LTR. During reverse transcription, RNase H degrades trailing RNA during RT, but the PPT resists degradation due to the abnormal stability of its RNA:DNA duplex52. The resistant RNA fragment primes plus-strand DNA synthesis. After plus-strand initiation, RT extends rightward through U3, R, U5, and PBS to produce plus-strand strong-stop DNA (+ssDNA), which then completes the second obligatory strand transfer to the 5′ end of the minus strand via PBS complementarity. The cPPT serves a similar internal priming function, and the two converging plus strands create the aforementioned central DNA flap.
SIN design: ΔU3 from the 3′ LTR is copied to both LTRs, silencing the provirus The 3′ LTR and the SIN deletion. The 3′ LTR retains HIV-1 U3–R–U5 but carries the self-inactivating (SIN) ΔU3 deletion: a ~400 bp removal of the U3 enhancer and promoter sequences, including NF-κB and Sp1 binding sites that normally drive HIV-1 transcription. Since reverse transcription copies the 3′ U3 to the 5′ LTR of the provirus, this deletion functionally silences both integrated LTRs to reduce undesired transcription of potentially recombined lentiviral genomic RNA or proximal genes. R in the 3′ LTR contains the active polyadenylation signal (PAS), where CPSF/CstF cleave to generate the poly(A). The terminal sequences of both LTRs contain the att sites—~15 bp inverted repeats (5′ TG...CA 3′) recognized by integrase for 3′ processing and strand transfer during integration. These are the only LTR sequences with a direct enzymatic function post-transduction; everything else in the LTR serves its purpose during transcription and reverse transcription and is functionally silent once integrated.
The three helper plasmids supply all trans-acting proteins. None contain Ψ or LTRs, strongly disfavoring packaging of their mRNAs (though the filter is not absolute: spliced viral and some host RNAs are measurably encapsidated at low frequency).
The Gag-Pol packaging plasmid encodes the Gag polyprotein (MA–CA–NC–p6) and, via −1 ribosomal frameshift at ~5% frequency, the Gag-Pol polyprotein (adding PR–RT–IN). MA targets Gag to PI(4,5)P2-rich membrane domains; CA forms the conical capsid core (~250 hexamers + 12 pentamers); NC’s zinc knuckles bind Ψ with nanomolar affinity; p6 recruits ESCRT (TSG101 via PTAP, ALIX via YPXnL) for budding. The mature virion contains ~4,000–5,000 Gag and ~120 Gag-Pol molecules, yielding ~50 RT heterodimers and ~100–150 IN molecules (consistent with Gag-Pol stoichiometry) inside the CA core53. Lentiviral capsids are 90–130 nm in diameter; comparatively, AAV capsids are ~25 nm.
The Rev plasmid supplies Rev, the only HIV-1 regulatory protein not engineered away. As previously mentioned, Rev oligomerizes on the RRE and recruits the CRM1/exportin-1 pathway to shuttle the unspliced vector RNA into the cytoplasm50. Without Rev, vector RNA is nuclear-localized and doesn’t reach Gag for packaging. Some groups have tried to replace Rev—Bray et al. substituted Mason-Pfizer monkey virus’ constitutive transport element (CTE), but at significant cost to titer51. In third-generation systems, Rev was separated onto its own plasmid (pRSV-Rev) to add a fourth independent recombination event required for any theoretical reconstitution of replication-competent virus, further improving biosafety3.
The VSV-G envelope plasmid replaces native HIV-1 Env (gp120–gp41) with the vesicular stomatitis virus glycoprotein, a highly consequential swap in lentiviral design. Wild-type HIV-1 Env uses a two-step entry mechanism: gp120 binds the primary receptor CD4, inducing a conformational change that exposes a co-receptor binding site for either CCR5 (R5-tropic, targeting macrophages and memory T cells) or CXCR4 (X4-tropic, targeting naïve T cells)42. This is ideal for in vivo pathogenesis but restricts tropism to CD4+ cells (preventing transduction of most common cell lines: HEK293T, HeLa, A549, U2OS, etc.). Additionally, Env expression in producer cells drives toxic syncytia formation (gp41-mediated cell–cell fusion that kills the cells). Env is notoriously difficult to express at high levels, poorly tolerant of concentration by ultracentrifugation, and shares extensive sequence homology with the packaging construct (increasing risk of recombination toward replication-competent lentivirus).
VSV-G entry: LDLR binding → endocytosis → pH fusion → core release VSV-G VSV is a negative-strand RNA rhabdovirus that infects cattle, horses, swine via arthropod vectors. VSV-G was first used to pseudotype retroviral vectors by Burns et al. (1993)44, who showed its broad receptor usage and resistance to ultracentrifugation could improve titer and tropism limitations of amphotropic retroviruses. Naldini et al. (1996)1 extended this to HIV-1-based lentiviral vectors, making it the default envelope ever since. VSV-G solves all of these problems. VSV-G binds the low-density lipoprotein receptor (LDLR) and related family members (LRP1, LRP2), which are ubiquitously expressed on most mammalian (and many non-mammalian) cell types, conferring versatile tropism (“pantropism”)43. Entry proceeds via clathrin-mediated endocytosis followed by pH-dependent membrane fusion in late endosomes (triggered at pH ~6.0–6.2), rather than at the plasma surface, meaning VSV-G pseudotyped virions don’t induce syncytia in producer cells44. VSV-G’s trimeric structure also tolerates ultracentrifugation at >50,000 × g, enabling ~300-fold concentration of viral supernatant without significant loss. Finally, VSV-G shares minimal homology with any HIV-1 component, reducing concerns for recombinational rescue of replication-competent virus.
VSV-G’s pantropism has exceptions, however. Quiescent (unstimulated) T cells, resting B cells, and hematopoietic stem and progenitor cells (HSPCs, CD34+) all express low levels of LDLR45. TCR stimulation or cytokine priming upregulates LDLR and rescues transduction, but forcing HSCs into cycle compromises their engraftment potential. These limitations have driven exploration of alternative envelope glycoproteins. Baboon endogenous retrovirus (BaEV) envelope, which enters via amino acid transporters ASCT-1 and ASCT-2, achieves ~80% transduction of TCR-stimulated T cells and 15–30% transduction of resting CD34+ HSCs without forcing cell-cycle entry46. Measles’ hemagglutinin and fusion glycoproteins (H/F), which bind SLAM and CD46, enabled the first efficient transduction of quiescent T and B cells47. Cocal virus glycoprotein (a VSV relative) offers comparable tropism to VSV-G but resists inactivation by human serum, an advantage for in vivo delivery and clinical manufacturing48. Rabies’ glycoprotein (RVG) pseudotypes exploit retrograde axonal transport, enabling transduction of motor neurons from non-invasive peripheral injection sites49. The envelope can thus be tailored to a target cell.
Standardized plasmids for each system are available from Addgene. For third-generation packaging: pMDLg/pRRE (Addgene #12251) supplies Gag-Pol, pRSV-Rev (Addgene #12253) expresses Rev; and pMD2.G (Addgene #12259) supplies VSV-G. For second-generation packaging, psPAX2 (Addgene #12260) encodes gag, pol, tat, and rev (combining the functions of pMDLg/pRRE and pRSV-Rev while retaining Tat). The envelope plasmid (pMD2.G) is shared across both systems. One example third-generation transfer plasmid is lentiGuide-Puro (Addgene #52963, Zhang lab), a widely used CRISPR sgRNA delivery backbone. One example second-generation transfer plasmid is pWPI (Addgene #12254, Trono lab), a bicistronic EF1α/IRES-EGFP expression vector. Notably, third-generation transfer plasmids are compatible with second-generation packaging plasmids, but second-generation transfer plasmids are incompatible with third-generation packaging plasmids (due to the absence of Tat). For an excellent practical overview of lentiviral vector biology, components, and protocols, see Addgene’s Lentiviral Vector Guide.
Packaging: From DNA to surface
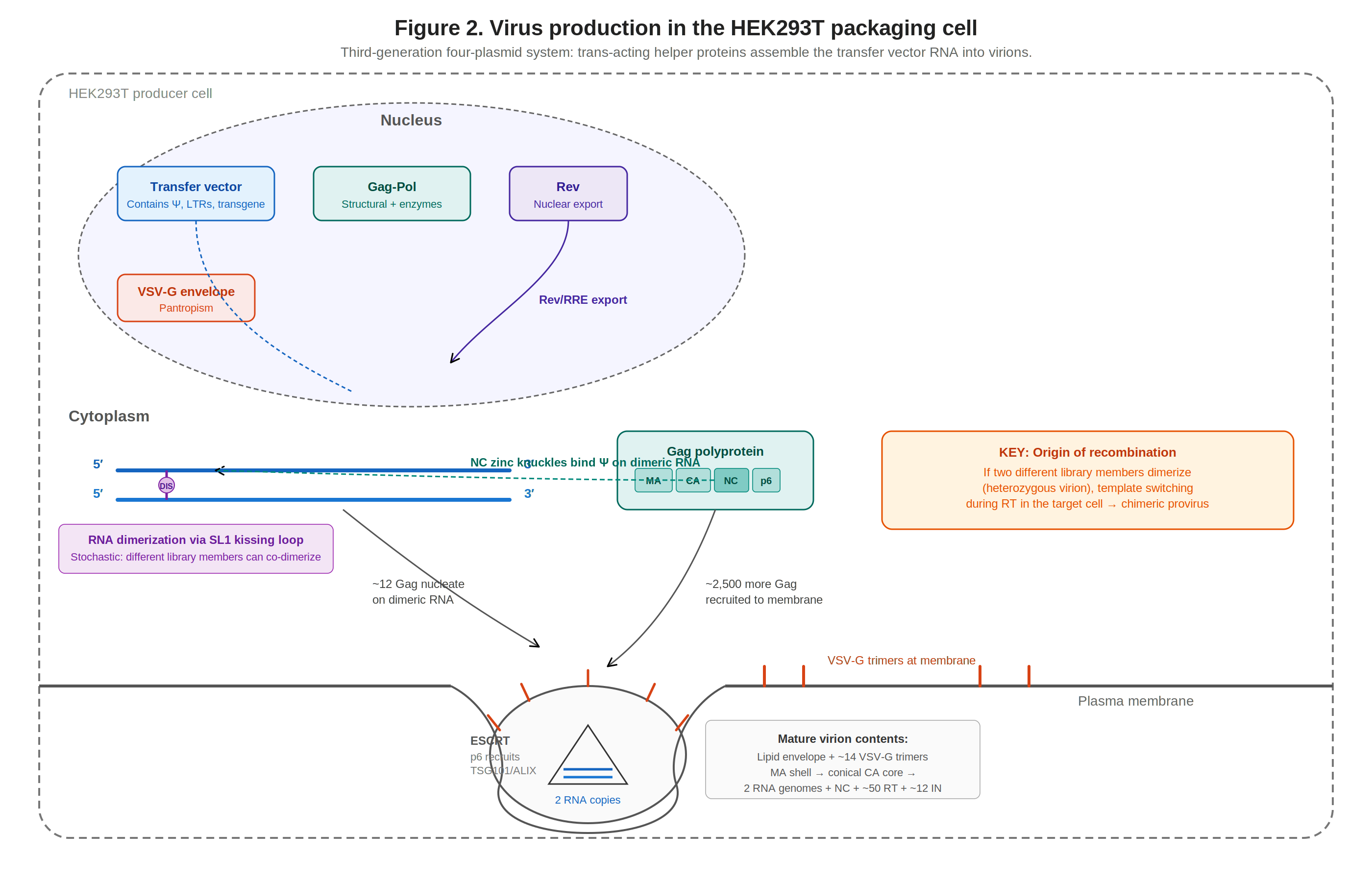
Figure 3. Virus production in HEK293T packaging cells. Four plasmids converge: transfer vector RNA dimerizes via DIS, is recognized by Gag-NC, and assembles into virions at the plasma membrane.
HEK293T is the standard packaging cell line for lentivirus. Unlike the parental HEK293 cell line, which was generated by transforming embryonic kidney cells with sheared adenovirus 5 DNA32, HEK293Ts were subsequently engineered from HEK293s for high expression of foreign genetic material by stable transfection with pRSV-1609, a plasmid encoding the SV40 virus’ large T antigen together with a neomycin phosphotransferase cassette (the resulting 293tsA1609neo line is the ancestor of modern HEK293T; see ATCC CRL-3216 and DuBridge et al., 1987, where the construct and line were originally described)33. As a byproduct, HEK293Ts are resistant to G418/neomycin (worth remembering when selecting selection strategies in HEK293Ts). This means HEK293Ts can also replicate episomes containing an SV40 origin of replication, which is present in many common expression vectors (including psPAX2, pMD2.G, and many transfer backbones). HEK293Ts’ high transfection efficiency (copy numbers can reach ~105–106 plasmid molecules per cell after lipofection, though only a poorly defined fraction is chromatinized into expression-competent templates57), tolerance of foreign DNA, and general robustness have established them as the standard for lentiviral packaging, producing over 10–100-fold more virus than parental HEK293s34.
Transfer vector RNA is transcribed from the chimeric 5′ LTR and exported via Rev/RRE. In the cytoplasm, vector RNAs dimerize via SL1’s DIS kissing-loop, and dimerized RNA is the form that predominantly ends up packaged. Worth noting, DIS initiates dimerization but is not strictly required for it: deleting or mutating the DIS palindrome reduces dimerization efficiency substantially but does not abolish it, and residual dimers still form through secondary contacts elsewhere in the 5′ UTR and gag region58. The mechanistic basis of this bias is not fully settled. One proposal is that the dimeric conformation exposes NC-binding sites that are partially occluded in the monomer, so Gag directly recognizes dimer-specific structural features; another is that Gag binds monomers and dimers with broadly similar affinity but oligomerizes/nucleates more efficiently on dimers because two juxtaposed Ψ elements accelerate the transition from initial binding to a productive assembly intermediate. These are not mutually exclusive, and the available SHAPE, CLIP, and in-cell cross-linking data are consistent with a mixture of both. What is well-established is the net outcome: dimeric RNA is preferentially packaged, providing a quality-control filter on which RNAs enter virions.
Mature lentiviral virion: conical CA core with diploid RNA inside a VSV-G-studded envelope Roughly ~8–12 Gag molecules are thought to nucleate on the dimeric RNA via NC54 (the exact number is not firmly pinned down), then traffic to the membrane where the rest of the lattice assembles, totaling ~4,000–5,000 Gag molecules per immature particle53. Host ESCRT-III catalyzes membrane scission. After budding, PR cleaves Gag/Gag-Pol, producing the mature virion: a lipid envelope studded with an estimated ~50 VSV-G trimers56 (extrapolated from native VSV measurements; no primary cryo-EM count on lentiviral pseudotypes has been published), an MA shell, and a conical CA core (~1,000–1,500 CA subunits arranged as ~250 hexamers + 12 pentamers) housing two RNA genomes plus NC, RT, and IN53. For pooled libraries, given the high plasmid copy number during vector production and the strong preference for dimeric RNA as the packaging substrate, many virions contain two different vector RNAs (heterozygous virions), providing the origin of inter-molecular recombination that can subsequently occur in transduced cells during reverse transcription.
Before transduction, it is common to titer viral supernatant or concentrated virus (both of which can be used for transduction). Typical methods to titer include p24 ELISA to measure CA (p24) concentration, flow cytometry to quantify fluorescence, qPCR to detect integration, and antibiotic selection to estimate the fraction of surviving cells. Commercial kits for p24 ELISA include Takara’s Lenti-X p24 Rapid Titer Kit (results within hours) and Lenti-X GoStix Plus, which uses a lateral-flow immunochromatographic strip to detect p24 in minutes (a companion smartphone app quantifies band intensity to estimate functional titer: a GoStix value of ~50 approximates 5 × 105 IFU/mL), although these are indirect proxies for functional titer.
Transduction: From surface to DNA
### Entry and Capsid Nuclear Import
VSV-G binds LDLR on the target cell surface, triggering clathrin-mediated endocytosis. At pH ~6.0–6.2, VSV-G undergoes a conformational change that drives membrane fusion, releasing the conical CA core into the cytoplasm.
The intact capsid core traverses the nuclear pore complex (NPC) narrow-end-first, engaging FG-nucleoporins (Nup358 at the cytoplasmic face, Nup153 at the nuclear basket). The NPC deforms to ~60 nm to accommodate the core. Cyclophilin A (CypA) in the cytoplasm blocks premature CPSF6 engagement; once in the nucleus, CPSF6 binds the CA pocket (the same pocket targeted by Gilead’s antiretroviral lenacapavir, a twice-yearly subcutaneous injection that achieved 100% efficacy—0 incident infections versus a background incidence of 2.41 per 100 person-years—in the PURPOSE-1 trial of 5,338 cisgender women in South Africa and Uganda55) and traffics the core to nuclear speckle-associated domains (SPADs). This capsid-mediated nuclear import mechanism is complex (getting nucleic acids from cytoplasm → nucleus is hard!) but is the fundamental reason lentiviruses can infect non-dividing cells, a capability gammaretroviral vectors lack (gammaretroviruses require mitotic nuclear envelope breakdown to access host DNA).
Reverse Transcription
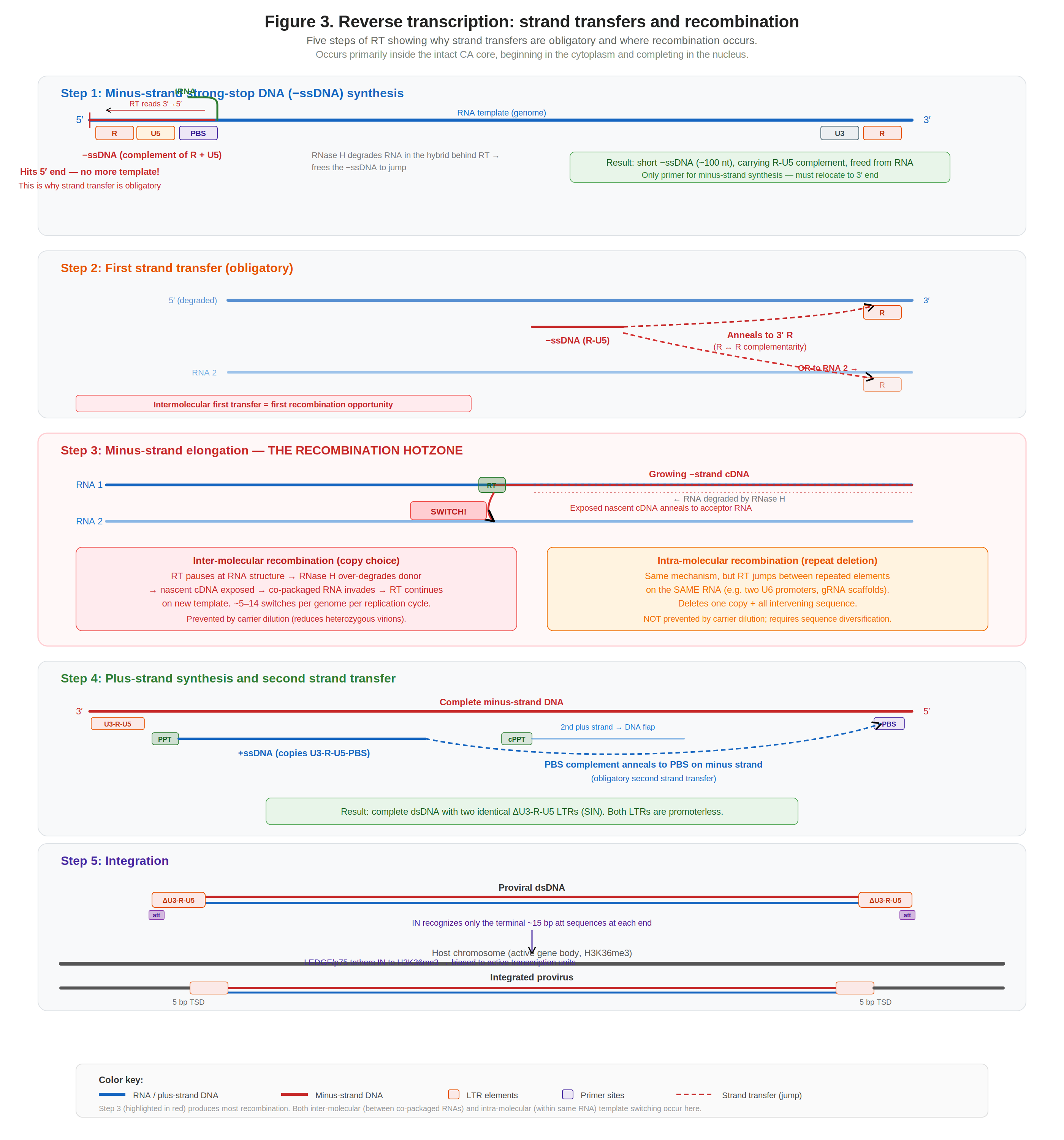
Figure 4. The five steps of reverse transcription, showing obligatory strand transfers and the recombination hotzone during minus-strand elongation (Step 3).
Reverse transcription converts the diploid RNA genome into a double-stranded DNA provirus ready for integration. Current evidence indicates RT starts in the cytoplasm but completes in the nucleus, within the intact CA core (a nanoscale reaction chamber concentrating RNA templates, RT, NC, and dNTPs imported through CA hexamer pores).
Step 1: Minus-strand strong-stop DNA synthesis. RT uses tRNALys3 at the PBS to initiate minus-strand synthesis, reading the template 3′→5′ (leftward toward the 5′ cap). It copies U5 and R (~100 nt), then hits the 5′ end; no more template. RNase H degrades the RNA in the hybrid, freeing the nascent −ssDNA.
Step 2: First strand transfer (obligatory). The freed −ssDNA jumps to the 3′ end of the genome by annealing its R complement to the 3′ R. This jump can occur intramolecularly (same RNA) or intermolecularly (co-packaged RNA): the first recombination opportunity. RT cannot initiate de novo; without this jump, reverse transcription stalls irreversibly.
Copy-choice recombination: RT pauses, RNase H degrades donor, cDNA switches to acceptor Step 3: Minus-strand elongation (the recombination hotzone). RT extends the minus strand along the entire genome from the 3′ R through U3, through the transgene cassette, all the way back toward the 5′ end. This is where the bulk of recombination occurs, via the dynamic copy-choice mechanism: RT pauses at RNA secondary structures, RNase H over-degrades the donor template ahead of the polymerase, the nascent cDNA is exposed, and the co-packaged acceptor RNA invades and anneals. An estimated 5–14 template-switching events occur per genome per cycle in HIV-114. As a useful heuristic, the recombination rate approximates ~1 crossover per kilobase of inter-element distance, though measured rates vary across constructs and contexts rather than behaving as a fixed per-kb constant.
Step 4: Plus-strand synthesis and second transfer. The PPT, an RNase H-resistant purine-rich RNA remnant, primes plus-strand DNA synthesis. RT copies U3-R-U5-PBS to produce +ssDNA, which jumps to the other end of the minus strand via PBS complementarity (second obligatory transfer). The cPPT provides a second priming site, and the two plus strands meet to form the central DNA flap.
Step 5: Integration. The completed dsDNA has two identical ΔU3-R-U5 LTRs (SIN). Integrase recognizes only the terminal ~15 bp att sequences at each end, performs 3′-processing (removing the terminal GT dinucleotide to expose the invariant CA-3′OH), and catalyzes strand transfer into host DNA with a 5 bp stagger. LEDGF/p75 tethers IN to H3K36me3 in active gene bodies, biasing integration into transcribed regions15.
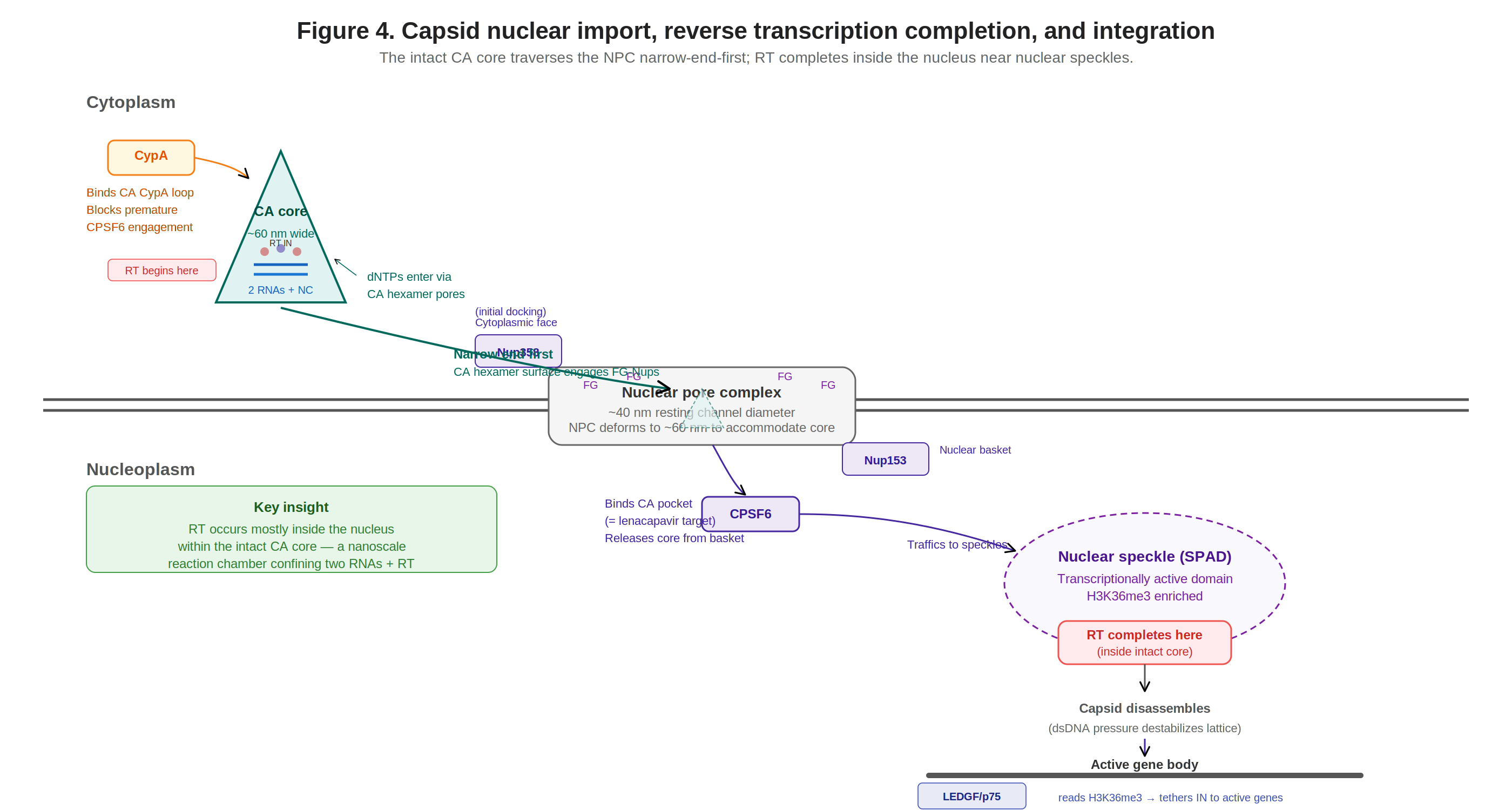
Figure 5. Capsid nuclear import, RT completion in the nucleus, and integration at nuclear speckle-associated domains via LEDGF/p75 tethering.
Determinants of Template-Switching Frequency
Polymerase domain mutations. Drug-resistance mutations (K65R, L74V, E89G, Q151N, M184I) slow polymerization and increase switching 2–6-fold. Hydroxyurea-mediated dNTP depletion increases switching ~1.8-fold, consistent with the model that pausing is the rate-limiting step for template switching.
RNase H mutations. H539N and D549N decrease switching ~2-fold18. More aggressive mutations (D443N, E478Q, D498N) further reduce switching but crash titer >1000-fold, making them impractical for vector production. Phenotypic mixing experiments in MLV showed a steady decline in repeat deletion frequency with decreasing functional RNase H, with >4-fold decreases when 95% of virion RT was RNase H-defective17.
Connection domain mutations. Patient-derived mutations (E312Q, G335C/D, N348I, A360I/V, V365I, A376S) reduce RNase H activity modestly while maintaining titer. In single-cycle assays, inclusion of these mutations reduced template-switching frequency from ~47% to ~32%16. N348I in particular maintains viral replication capacity, reduces secondary RNase H cleavages, and increases processive DNA synthesis, an attractive combination for vector engineering. Combining N348I with A360V and a modest RNase H site mutation (D549N) might achieve ~3-fold reduction in switching while keeping titer usable.
Nuclear dNTP pools (speculative). Because RT appears to complete in the nucleus in many settings, the relevant dNTP concentration for the late stages of synthesis may be the nuclear pool rather than the cytoplasmic pool. Nuclear dNTP levels are generally reported as lower than cytoplasmic levels, which would be expected to favor RT pausing and template switching. This raises the possibility that the nuclear environment itself contributes to the baseline recombination rate, independent of heterozygous virion frequency, but this is a hypothesis rather than an established finding and the quantitative contribution has not been measured directly.
Recombination as a Problem for Pooled Screens
The earliest genome-wide loss-of-function libraries used shRNA hairpins delivered by lentivirus. Stable integration meant each cell carried a heritable, barcode-like perturbation that could be tracked by deep sequencing after phenotypic selection, a property no transient delivery method could match. When CRISPR-Cas9 arrived, the same lentiviral infrastructure was repurposed almost overnight: the shRNA cassette was swapped for a guide-RNA expression cassette, and the first genome-scale CRISPR knockout screens45 were published within months of each other. This made lentiviral vectors the workhorse of pooled genetic screens. The platform rapidly expanded to CRISPRi/CRISPRa transcriptional control67, single-cell transcriptomic readouts that pair perturbation identity with gene expression (Perturb-seq8; CROP-seq9), and ultimately genome-scale perturbation profiling of over one million cells in a single experiment10.
In every case, the lentiviral vector is the common thread: it integrates a single perturbation per cell, maintains it through division, and encodes a sequenceable identifier. But the same retroviral biology that enables this efficient delivery introduces an intrinsic source of potential noise: genetic recombination during reverse transcription.
Inter-molecular Recombination: The Perturb-seq Problem
Inter-molecular recombination occurs when RT switches templates between co-packaged RNAs in heterozygous virions. This was the central technical challenge of the original Perturb-seq platform. The Dixit et al. (2016) vector placed the sgRNA (mU6 promoter) and a polyadenylated guide barcode (GBC) in antiparallel cassettes ~2.7 kb apart8. When the library was pooled prior to virus production, template switching during reverse transcription scrambled sgRNA–GBC linkages at high frequency.
Xie et al. (2018) subsequently quantified this directly in Mosaic-seq libraries: when libraries were pooled before viral packaging, the most abundant sgRNA for each barcode occupied a median of only ~42% of reads, meaning >50% of sgRNA–barcode linkages were scrambled19. In contrast, individually packaged viruses maintained >83% correct linkage. This confirmed that recombination occurs overwhelmingly during reverse transcription of co-packaged heterodimeric genomes, not during co-infection of the same cell by independent virions.
The companion Adamson et al. (2016) study anticipated this problem and mitigated it through arrayed cloning: each sgRNA–GBC pair was individually cloned and verified by Sanger sequencing before pooling, creating a known dictionary that allowed computational identification of recombinant proviruses20. This preserved sgRNA–GBC fidelity but was laborious and not scalable to genome-wide libraries. Their subsequent 2018 preprint provided detailed best practices and discussed three mitigation strategies: arrayed library preparation, carrier plasmid dilution, and the CROP-seq architecture.
The most systematic recent measurement comes from the Blainey lab's CROPseq-multi study, which assayed recombination across eight plasmid libraries and four cell lines12. They observed 9–17% total recombination (plasmid + lentiviral), with a mean of 12% attributable to lentiviral integration. Recombination frequency scaled approximately linearly with inter-element distance, consistent in magnitude with the ~1 crossover/kb heuristic from the virology literature, though the measured per-kb rate varied across constructs and contexts rather than landing on a single universal value.
Intra-molecular Recombination: The Multi-Guide Problem
Intra-molecular recombination is mechanistically distinct: RT jumps between repeated sequence elements on the same RNA molecule, deleting one copy and all intervening sequence. This cannot be prevented by carrier dilution, since it occurs on a single genome.
The shRNA gene therapy precedent. The problem was first characterized extensively in the HIV-1 gene therapy field, where multiple shRNAs are needed simultaneously to prevent viral escape. ter Brake et al. (2008) tested multi-shRNA vectors with repeated H1 or U6 promoters and found that identical promoters caused frequent cassette deletion21. In triple-cassette vectors with identical promoters, only 13% of SupT1 cell clones retained the intact construct. A comprehensive study by McIntyre et al. (2009) generated >500 clonal cell lines with 2–6 repeated shRNA cassettes and found deletion frequencies ranging from 2% to 36%, with central positions deleted most frequently22.
The TALEN disaster. TALEN DNA-binding domains consist of ~34 amino acid repeats with ~97% sequence identity at the nucleotide level. Mock et al. (2014) showed that lentiviral vectors package full-length TALEN mRNAs intact, but 100% of single-cell clones (25/25) showed recombination in the repeat domain after reverse transcription23. In 39 of 40 analyzed clones, recombination occurred between identical nucleotides of different repeats, resulting in in-frame elimination of complete repeat modules. This was so severe that the group developed RT-dead lentiviral particles (NRTLVs) for mRNA-only delivery as a workaround. The TALEN case represents the extreme of what happens when high-identity tandem repeats meet lentiviral reverse transcription.
Dual-gRNA vectors for CRISPR screens. Vidigal and Ventura (2015) directly demonstrated the problem: lentiviral vectors expressing gRNA pairs from two identical hU6 promoters lost the proximal gRNA cassette, confirmed by genomic PCR24. Replacing one hU6 with a synthetic murine U6 variant eliminated recombination. Vendor-reported data from VectorBuilder (2024, technical note) illustrate the same trend quantitatively: a 249 bp duplicated stuffer reportedly reduced upstream reporter expression from 96% to 19% of cells, and identical CMV promoters (568 bp) left only ~12% expressing the upstream reporter25. These numbers should be read as vendor benchmarking rather than primary literature, and are consistent with the broader observation that homology length and homology identity are the dominant determinants of intramolecular template switching.
Recombination during reverse transcription is not a rare exception but a systematic constraint. It affects the design of every lentiviral construct, and the cost of ignoring it scales with library complexity.
Mitigation Strategies
Reducing Heterozygous Virion Formation
Carrier plasmid dilution. Feldman et al. (2018) demonstrated that co-transfecting a 1:100 library-to-carrier transfer vector ratio ensures >99% of library-containing virions are paired with carrier RNA, not another library member26. This nearly eliminates inter-molecular recombination but reduces effective titer ~100-fold, requiring scaled-up virus production. The approach has been used successfully in multiple Perturb-seq studies.
DIS subtype incompatibility. Chen et al. (2009) showed that replacing the DIS palindrome (subtype B: GCGCGC) with subtype C (GUGCAC) reduces cross-pool co-packaging ~9-fold27. This approach is limited by the small number of validated orthogonal DIS classes.
Eliminating Intra-molecular Repeats
Orthogonal promoters. The standard approach since ter Brake et al. (2008). Kabadi et al. (2014) assembled four sgRNAs under hU6, mU6, 7SK, and H1 via Golden Gate cloning28. Limited by the small number of compact Pol III promoters with validated gRNA expression.
Orthogonal scaffolds. The Adamson et al. (2016) vector used modified gRNA constant regions (cr2, cr3 variants) alongside distinct promoters. CROPseq-multi extends this with tRNA-processed arrays using orthogonal scaffolds12.
Dual-nuclease systems. CHyMErA pairs SpCas9 with Cas12a, which uses a completely different guide RNA structure, inherently eliminating all scaffold homology13. Benchmarking by Najm et al. (2018) across ten digenic CRISPR technologies found that alternative tracrRNA sequences from SpCas9 consistently showed superior performance for combinatorial screens29.
Minimizing inter-element distance. The Big Papi vector uses antiparallel promoters to place spacers <200 bp apart, reducing recombination to ~9% (CROPseq-multi measurements). Cas12a systems with native crRNA array processing separate spacers by only ~20 bp, approaching negligible recombination.
Readout-Level Solutions
CROP-seq. Datlinger et al. (2017) placed the sgRNA cassette in the 3′ LTR9. During provirus synthesis, LTR duplication copies the sgRNA to both ends, producing a Pol II-driven copy that can be captured directly in scRNA-seq. Because the sgRNA itself is the barcode, recombination between sgRNA and a distant barcode is irrelevant. This design has largely supplanted the original Perturb-seq architecture.
Direct-capture Perturb-seq. Replogle et al. (2020) developed modified sgRNA scaffolds with capture sequences enabling direct detection of multiple sgRNAs per cell, supporting combinatorial screens without barcodes.
Computational filtering. The Replogle et al. (2022) genome-scale CRISPRi Perturb-seq used direct guide capture, largely bypassing the recombination problem10. The Horlbeck/Replogle dual-sgRNA CRISPRi library accepted ~20–30% recombination and computationally removed affected cells, increasing required cell numbers proportionally.
Alternative Delivery Systems
Transposon-based delivery. PiggyBac and Sleeping Beauty eliminate all RT-mediated recombination entirely. Limited by delivery efficiency (transfection/electroporation) and MOI control. Used for CRISPR screens in specific contexts but have not displaced lentiviral delivery broadly.
Non-reverse-transcribable LVs (NRTLVs). Mock et al. (2014) inactivated RT entirely for mRNA-only delivery of TALENs23. Incompatible with pooled screens requiring stable integration.
Gag-only VLPs. Recent systems (Haldrup et al., 2023; Jia et al., 2025) eliminate both RT and IN, packaging CRISPR RNPs or base editor mRNA. Useful for hit-and-run editing, not for screens requiring stable proviral barcodes.
The Unexplored Frontier: RT Engineering
The most mechanistically direct but least explored approach to reducing recombination is to modify reverse transcriptase itself. The virology literature provides extensive characterization of mutations that modulate recombination, but none have been tested in a lentiviral packaging plasmid for screening applications.
The connection domain mutations offer the most promising starting point. N348I alone reduced template switching from ~47% to ~32% in the GFP reconstitution assay while maintaining viral replication capacity16. Ehteshami et al. (2008) further characterized A360V as reducing secondary RNase H cleavages through both RNase H-dependent and -independent mechanisms30. These mutations were identified in the context of AZT resistance, but their recombination-reducing phenotype makes them attractive for vector engineering. Combining N348I with A360V and a modest RNase H site mutation (D549N) could plausibly achieve ~3-fold reduction in switching while keeping titer usable.
The gap between virology and functional genomics is striking. Despite decades of RT characterization, no group has built a packaging plasmid carrying these mutations and tested it for library delivery. The screening community treats the packaging plasmid as a black box. We estimate that a systematic effort: testing 5–10 RT mutant combinations for titer, recombination frequency, and screen performance, would require approximately 3–6 months of focused work and could quantify the practical benefit of RT engineering.
These effects should be additive with carrier dilution (which addresses inter-molecular events orthogonally) and with orthogonal vector elements (which address intra-molecular events at the repeat level). A combined strategy (RT mutations + carrier dilution + orthogonal cassette design) could in principle reduce total recombination to low single-digit percentages, a regime where computational filtering becomes nearly costless.
Summary and Future Directions
Lentiviral recombination in pooled screens emerges from the fundamental biology of the vector system: RNA dimerization is coupled to packaging, diploid genomes are the norm, and template switching is an intrinsic property of RT that serves essential functions (obligatory strand transfers) alongside generating unwanted crossovers. Two decades of screening experience have produced a toolkit of workarounds: carrier dilution, CROP-seq, orthogonal promoters/scaffolds, dual-nuclease systems, and computational filtering. These have been sufficient for most single-guide screen designs.
However, as the field moves toward combinatorial screens, multi-modal readouts, and higher-order perturbation arrays, the cost of recombination increases. Intra-molecular recombination in particular, which is not addressed by carrier dilution, imposes an increasingly severe constraint on vector design. The architectural solutions (orthogonal promoters, minimized inter-element distance) are approaching their physical limits: there are only a handful of validated compact Pol III promoters, and Cas12a systems, while elegant, lag in guide design tools and perturbation modality breadth.
The most promising and least explored direction is systematic RT engineering in the packaging plasmid. The virology and functional genomics communities would benefit from closer collaboration on this front. The mutations are characterized, the packaging plasmids are well-defined, and the assays (single-cycle GFP reconstitution, paired sgRNA sequencing after transduction) are established. What is missing is the systematic engineering effort to bridge these two fields.
References
- Naldini, L., et al. (1996). In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science, 272(5259), 263–267. doi:10.1126/science.272.5259.263 ↑
- Zufferey, R., et al. (1997). Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nature Biotechnology, 15(9), 871–875. doi:10.1038/nbt0997-871 ↑
- Dull, T., et al. (1998). A third-generation lentivirus vector with a conditional packaging system. Journal of Virology, 72(11), 8463–8471. doi:10.1128/JVI.72.11.8463-8471.1998 ↑
- Shalem, O., et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science, 343(6166), 84–87. doi:10.1126/science.1247005 ↑
- Wang, T., et al. (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Science, 343(6166), 80–84. doi:10.1126/science.1246981 ↑
- Gilbert, L.A., et al. (2014). Genome-scale CRISPR-mediated control of gene repression and activation. Cell, 159(3), 647–661. doi:10.1016/j.cell.2014.09.029 ↑
- Horlbeck, M.A., et al. (2016). Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife, 5, e19760. doi:10.7554/eLife.19760 ↑
- Dixit, A., et al. (2016). Perturb-Seq: Dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell, 167(7), 1853–1866. doi:10.1016/j.cell.2016.11.038 ↑
- Datlinger, P., et al. (2017). Pooled CRISPR screening with single-cell transcriptome readout. Nature Methods, 14(3), 297–301. doi:10.1038/nmeth.4177 ↑
- Replogle, J.M., et al. (2022). Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq. Cell, 185(14), 2559–2575. doi:10.1016/j.cell.2022.05.013 ↑
- Zennou, V., et al. (2000). HIV-1 genome nuclear import is mediated by a central DNA flap. Cell, 101(2), 173–185. doi:10.1016/S0092-8674(00)80828-4; Follenzi, A., et al. (2000). Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV-1 pol sequences. Nature Genetics, 25(2), 217–222. doi:10.1038/76095 ↑
- Walton, R.T., et al. (2025). CROPseq-multi: a universal solution for multiplexed perturbation in high-content pooled CRISPR screens. bioRxiv. doi:10.1101/2024.03.17.585235 ↑
- Gonatopoulos-Pournatzis, T., et al. (2020). Genetic interaction mapping and exon-resolution functional genomics with a hybrid Cas9–Cas12a platform. Nature Biotechnology, 38, 638–648. doi:10.1038/s41587-020-0437-z ↑
- Levy, D.N., et al. (2004). Dynamics of HIV-1 recombination in its natural target cells. PNAS, 101(12), 4204–4209. doi:10.1073/pnas.0306764101 ↑
- Ciuffi, A., et al. (2005). A role for LEDGF/p75 in targeting HIV DNA integration. Nature Medicine, 11(12), 1287–1289. doi:10.1038/nm1329 ↑
- Nikolenko, G.N., et al. (2007). Mutations in the connection domain of HIV-1 reverse transcriptase increase 3′-azido-3′-deoxythymidine resistance. PNAS, 104(1), 317–322. doi:10.1073/pnas.0609642104 ↑
- Hwang, C.K., Svarovskaia, E.S. & Pathak, V.K. (2001). Dynamic copy choice: steady state between murine leukemia virus polymerase and polymerase-dependent RNase H activity determines frequency of in vivo template switching. PNAS, 98(21), 12209–12214. doi:10.1073/pnas.221289898 ↑
- Nikolenko, G.N., et al. (2005). Mechanism for nucleoside analog-mediated abrogation of HIV-1 replication: balance between RNase H activity and nucleotide excision. PNAS, 102(6), 2093–2098. doi:10.1073/pnas.0409823102 ↑
- Xie, S., et al. (2018). Frequent sgRNA-barcode recombination in single-cell perturbation assays. PLoS ONE, 13(6), e0198635. doi:10.1371/journal.pone.0198635 ↑
- Adamson, B., et al. (2016). A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell, 167(7), 1867–1882. doi:10.1016/j.cell.2016.11.048 ↑
- ter Brake, O., et al. (2008). Lentiviral vector design for multiple shRNA expression and durable HIV-1 gene therapy. Molecular Therapy, 16(3), 557–564. doi:10.1038/sj.mt.6300382 ↑
- McIntyre, G.J., et al. (2009). Cassette deletion in multiple shRNA lentiviral vectors for HIV-1 and its impact on treatment success. Virology Journal, 6, 184. doi:10.1186/1743-422X-6-184 ↑
- Mock, U., et al. (2014). Novel lentiviral vectors with mutated reverse transcriptase for mRNA delivery of TALE nucleases. Scientific Reports, 4, 6409. doi:10.1038/srep06409 ↑
- Vidigal, J.A. & Ventura, A. (2015). Rapid and efficient one-step generation of paired gRNA CRISPR-Cas9 libraries. Nature Communications, 6, 8083. doi:10.1038/ncomms9083 ↑
- VectorBuilder (2024). Technical note: lentiviral recombination with duplicated sequences. vectorbuilder.com ↑
- Feldman, D., Singh, A., Garrity, A.J. & Blainey, P.C. (2018). Lentiviral co-packaging mitigates the effects of intermolecular recombination and multiple integrations in pooled genetic screens. bioRxiv. doi:10.1101/262121 ↑
- Chen, J., et al. (2009). High efficiency of HIV-1 genomic RNA packaging and heterozygote formation revealed by single virion analysis. PNAS, 106(32), 13535–13540. doi:10.1073/pnas.0906822106 ↑
- Kabadi, A.M., et al. (2014). Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Research, 42(19), e147. doi:10.1093/nar/gku749 ↑
- Najm, F.J., et al. (2018). Orthologous CRISPR–Cas9 enzymes for combinatorial genetic screens. Nature Biotechnology, 36, 179–189. doi:10.1038/nbt.4048 ↑
- Ehteshami, M., et al. (2008). Connection domain mutations N348I and A360V in HIV-1 reverse transcriptase enhance resistance to 3′-azido-3′-deoxythymidine through both RNase H-dependent and -independent mechanisms. Journal of Biological Chemistry, 283(32), 22222–22232. doi:10.1074/jbc.M803521200 ↑
- Vink, C.A., et al. (2017). Eliminating HIV-1 packaging sequences from lentiviral vector proviruses enhances safety and expedites gene transfer for gene therapy. Molecular Therapy, 25(8), 1790–1804. doi:10.1016/j.ymthe.2017.04.028 ↑
- Graham, F.L., et al. (1977). Characteristics of a human cell line transformed by DNA from human adenovirus type 5. Journal of General Virology, 36(1), 59–72. doi:10.1099/0022-1317-36-1-59 ↑
- ATCC. 293T/17 [HEK 293T/17], CRL-3216, lineage and genotype documentation. atcc.org/products/crl-3216; DuBridge, R.B., Tang, P., Hsia, H.C., Leong, P.-M., Miller, J.H. & Calos, M.P. (1987). Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Molecular and Cellular Biology, 7(1), 379–387. doi:10.1128/mcb.7.1.379-387.1987 (original description of the pRSV-1609 / 293tsA1609neo construct). ↑
- Pear, W.S., et al. (1993). Production of high-titer helper-free retroviruses by transient transfection. PNAS, 90(18), 8392–8396. doi:10.1073/pnas.90.18.8392 ↑
- Lee, S. & Cobrinik, D. (2020). Improved third-generation lentiviral packaging with pLKO.1C vectors. BioTechniques, 68(6), 349–352. doi:10.2144/btn-2019-0155 ↑
- Malim, M.H. & Emerman, M. (2008). HIV-1 accessory proteins—ensuring viral survival in a hostile environment. Cell Host & Microbe, 3(6), 388–398. doi:10.1016/j.chom.2008.04.008 ↑
- Sheehy, A.M., et al. (2002). Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature, 418(6898), 646–650. doi:10.1038/nature00939 ↑
- Laguette, N., et al. (2014). Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell, 156(1–2), 134–145. doi:10.1016/j.cell.2013.12.011 ↑
- Neil, S.J., et al. (2008). Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature, 451(7177), 425–430. doi:10.1038/nature06553 ↑
- Kirchhoff, F. (2010). Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host & Microbe, 8(1), 55–67. doi:10.1016/j.chom.2010.06.004 ↑
- Samson, M., et al. (1996). Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature, 382(6593), 722–725. doi:10.1038/382722a0 ↑
- Berger, E.A., Murphy, P.M. & Farber, J.M. (1999). Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annual Review of Immunology, 17, 657–700. doi:10.1146/annurev.immunol.17.1.657 ↑
- Finkelshtein, D., et al. (2013). LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. PNAS, 110(18), 7306–7311. doi:10.1073/pnas.1214441110 ↑
- Burns, J.C., et al. (1993). Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. PNAS, 90(17), 8033–8037. doi:10.1073/pnas.90.17.8033 ↑
- Amirache, F., et al. (2014). Mystery solved: VSV-G-LVs do not allow efficient gene transfer into unstimulated T cells, B cells, and HSCs because they lack the LDL receptor. Blood, 123(9), 1422–1424. doi:10.1182/blood-2013-11-540906 ↑
- Girard-Gagnepain, A., et al. (2014). Baboon envelope pseudotyped LVs outperform VSV-G-LVs for gene transfer into early-cytokine-stimulated and resting HSCs. Blood, 124(8), 1221–1231. doi:10.1182/blood-2014-02-558163 ↑
- Frecha, C., et al. (2008). Stable transduction of quiescent T cells without induction of cycle progression by a novel lentiviral vector pseudotyped with measles virus glycoproteins. Blood, 112(13), 4843–4852. doi:10.1182/blood-2008-05-155945 ↑
- Trobridge, G.D., et al. (2010). Cocal-pseudotyped lentiviral vectors resist inactivation by human serum and efficiently transduce primate hematopoietic repopulating cells. Molecular Therapy, 18(4), 725–733. doi:10.1038/mt.2009.282 ↑
- Hislop, J.N., et al. (2014). Rabies virus envelope glycoprotein targets lentiviral vectors to the axonal retrograde pathway in motor neurons. Journal of Biological Chemistry, 289(23), 16148–16163. doi:10.1074/jbc.M114.549980 ↑
- Pollard, V.W. & Malim, M.H. (1998). The HIV-1 Rev protein. Annual Review of Microbiology, 52, 491–532. doi:10.1146/annurev.micro.52.1.491 ↑
- Bray, M., et al. (1994). A small element from the Mason-Pfizer monkey virus genome makes human immunodeficiency virus type 1 expression and replication Rev-independent. PNAS, 91(4), 1256–1260. doi:10.1073/pnas.91.4.1256 ↑
- Lesnik, E.A. & Freier, S.M. (1995). Relative thermodynamic stability of DNA, RNA, and DNA:RNA hybrid duplexes: relationship with base composition and structure. Biochemistry, 34(34), 10807–10815. doi:10.1021/bi00034a013; Sugimoto, N., et al. (1995). Thermodynamic parameters to predict stability of RNA/DNA hybrid duplexes. Biochemistry, 34(35), 11211–11216. doi:10.1021/bi00035a029 ↑
- Briggs, J.A.G., et al. (2004). The stoichiometry of Gag protein in HIV-1. Nature Structural & Molecular Biology, 11(7), 672–675. doi:10.1038/nsmb785; Carlson, L.-A., et al. (2008). Three-dimensional analysis of budding sites and released virus suggests a revised model for HIV-1 morphogenesis. Cell Host & Microbe, 4(6), 592–599. doi:10.1016/j.chom.2008.10.013 ↑
- Kutluay, S.B. & Bieniasz, P.D. (2010). Analysis of the initiating events in HIV-1 particle assembly and genome packaging. PLoS Pathogens, 6(11), e1001200. doi:10.1371/journal.ppat.1001200 ↑
- Bekker, L.-G., et al. (2024). Twice-Yearly Lenacapavir or Daily F/TAF for HIV Prevention in Cisgender Women. New England Journal of Medicine, 391(13), 1179–1192. doi:10.1056/NEJMoa2407001; Kelley, C.F., et al. (2025). Twice-Yearly Lenacapavir for HIV Prevention in Men and Gender-Diverse Persons. New England Journal of Medicine, 392(13), 1261–1276. doi:10.1056/NEJMoa2411858 ↑
- Thomas, D., et al. (1985). Mass and molecular composition of vesicular stomatitis virus: a scanning transmission electron microscopy analysis. Journal of Virology, 54(2), 598–607. doi:10.1128/jvi.54.2.598-607.1985. (Native VSV virions carry ~1,205 G protein monomers, i.e. ~400 trimers; VSV-G pseudotyped lentiviral particles, lacking the VSV M-protein scaffold and built on a smaller spherical envelope, incorporate substantially fewer—on the order of ~50 trimers per particle—though no primary cryo-EM enumeration on lentiviral pseudotypes has been published.) ↑
- Cohen, R.N., et al. (2009). Quantification of plasmid DNA copies in the nucleus after lipoplex and polyplex transfection. Journal of Controlled Release, 135(2), 166–174. doi:10.1016/j.jconrel.2008.12.016; Glover, D.J., et al. (2005). Towards safe, non-viral therapeutic gene expression in humans. Nature Reviews Genetics, 6(4), 299–310. doi:10.1038/nrg1577 ↑
- Berkhout, B. & van Wamel, J.L. (1996). Role of the DIS hairpin in replication of human immunodeficiency virus type 1. Journal of Virology, 70(10), 6723–6732. doi:10.1128/jvi.70.10.6723-6732.1996; Clever, J.L. & Parslow, T.G. (1997). Mutant human immunodeficiency virus type 1 genomes with defects in RNA dimerization or encapsidation. Journal of Virology, 71(5), 3407–3414. doi:10.1128/jvi.71.5.3407-3414.1997 ↑
Notes
- vpu also shares a bicistronic mRNA with env and initiates from a non-canonical ACG start codon in subtype D isolates. There are nine common HIV-1 subtypes, with subtype C comprising approximately 50% of global cases. ↑
- CD4 is the primary receptor for wt HIV-1 entry; gp120 binding exposes a co-receptor site, and engagement of CCR5 or CXCR4 triggers gp41-mediated membrane fusion. CCR5 is the same gene disrupted by He Jiankui in the 2018 germline-editing experiment on twin embryos, an attempt to confer HIV resistance based on the naturally occurring CCR5-Δ32 deletion found in ~1% of Europeans41. ↑
- HIV-1 DIS kissing-loops have been programmably co-opted to form condensates of RNA nanostars annealed to one another. Fabrini et al. (2024, Nature Nanotechnology 19, 1665–1673) designed four-armed RNA nanostars whose arms terminated in kissing-loop sequences derived from the HIV-1 DIS, and visualized micron-scale RNA condensate formation via fluorescent light-up aptamers (like Broccoli) that fluoresce upon interaction with small-molecule fluorophore mimics. ↑
- VSV is a negative-strand RNA rhabdovirus that infects cattle, horses, swine via arthropod vectors. VSV-G was first used to pseudotype retroviral vectors by Burns et al. (1993)44, who showed its broad receptor usage and resistance to ultracentrifugation could improve titer and tropism limitations of amphotropic retroviruses. Naldini et al. (1996)1 extended this to HIV-1-based lentiviral vectors, making it the default envelope ever since. ↑